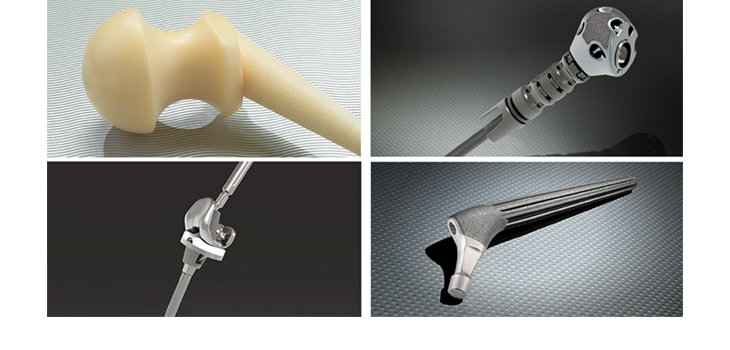
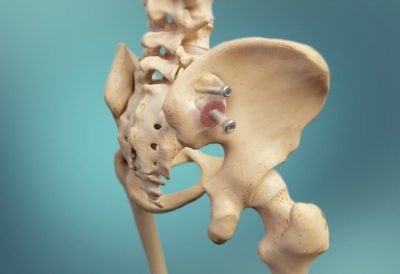
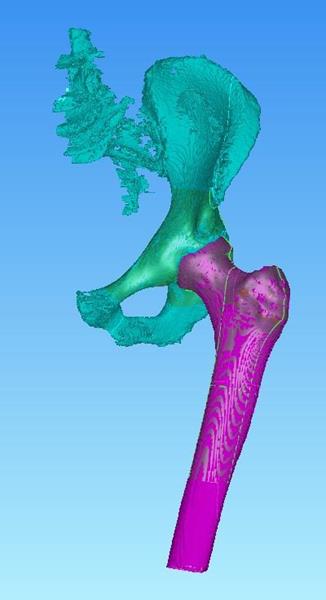
LEESBURG, Va., June 22, 2017 (GLOBE NEWSWIRE) — K2M Group Holdings, Inc. (NASDAQ:KTWO) (the “Company” or “K2M”), a global leader of complex spine and minimally invasive solutions focused on achieving three-dimensional Total Body Balance™, today announced that its MOJAVE™ PL 3D Expandable Interbody System has received 510(k) clearance from the U.S. Food & Drug Administration (FDA). MOJAVE PL 3D is a first-to-market, FDA-cleared, 3D-printed expandable posterior-lumbar (PL) interbody system that features K2M’s Lamellar 3D Titanium Technology™. K2M was the first leading spine company to market a 3D-printed titanium interbody device and offers the most comprehensive portfolio of 3D-printed spinal devices.
The MOJAVE PL 3D Expandable Interbody System is a fusion device designed to allow for independent control of the anterior and posterior height in the lumbar spine, a new capability not available with any other product in the market today. Featuring infinite adjustment within the expansion range, the implant may be locked at any desired height and lordosis to aid in the restoration of sagittal balance.
“The ability to provide independent control of both the anterior and posterior height separately is desirable for the restoration of sagittal balance compared to existing devices that can’t independently adjust both anterior and posterior height,” said Steven Ludwig, MD, an orthopedic spine surgeon and professor of orthopedics at the University of Maryland Medical Center in Baltimore.
K2M’s Lamellar 3D Titanium Technology uses an advanced 3D printing method to create structures that are impossible with traditional manufacturing techniques. Starting with a titanium powder, the MOJAVE PL 3D endplates are grown through the selective application of a high-energy laser beam, incorporating complex internal geometries and rough surface architecture that pre-clinical data have associated with bone growth activity. Lamellar 3D Titanium Technology incorporates a porous structure along with rough surfaces to allow the potential for bony integration throughout the endplates.
“We are proud to be the global leader in 3D printing of spinal applications. We have developed internal 3D expertise that is allowing us to accelerate the rate of spinal innovation. As the first-ever, FDA-cleared, 3D-printed expandable interbody technology, MOJAVE PL 3D exemplifies our leadership in this space and provides surgeons the ability to expand the implant in-situ. This is our second family of products featuring Lamellar 3D Titanium Technology and builds upon the incredibly successful CASCADIA™ 3D family of static 3D-printed interbody cages,” said K2M President and CEO Eric Major. “Our continued innovation in 3D solutions and our focus on 3D spinal balance, demonstrated by the recent launch of our Balance ACS™ platform, solidifies our position as a market leader and innovator in the industry. We look forward to continuing to introduce new three-dimensional solutions to surgeons who treat patients with spinal disorders.”
Balance ACS (BACS™) is a comprehensive platform applying three-dimensional solutions across the entire clinical care continuum to help drive quality outcomes in spine patients. BACS provides solutions focused on achieving balance of the spine by addressing each anatomical vertebral segment with a 360-degree approach to the axial, coronal, and sagittal planes, emphasizing Total Body Balance as an important component of surgical success.
For more information about the MOJAVE PL 3D Expandable Interbody System and Lamellar 3D Titanium Technology, visit www.K2M.com. For more information about K2M’s Balance ACS platform, visit www.BACS.com.
About K2M
K2M Group Holdings, Inc. is a global leader of complex spine and minimally invasive solutions focused on achieving three-dimensional Total Body Balance. Since its inception, K2M has designed, developed, and commercialized innovative complex spine and minimally invasive spine technologies and techniques used by spine surgeons to treat some of the most complicated spinal pathologies. K2M has leveraged these core competencies into Balance ACS, a platform of products, services, and research to help surgeons achieve three-dimensional spinal balance across the axial, coronal, and sagittal planes, with the goal of supporting the full continuum of care to facilitate quality patient outcomes. The Balance ACS platform, in combination with the Company’s technologies, techniques, and leadership in the 3D-printing of spinal devices, enable K2M to compete favorably in the global spinal surgery market. For more information, visit www.K2M.com and connect with us on Facebook, Twitter, Instagram, LinkedIn, and YouTube.
Forward-Looking Statements
This press release contains forward-looking statements that reflect current views with respect to, among other things, operations and financial performance. Forward-looking statements include all statements that are not historical facts such as our statements about our expected financial results and guidance and our expectations for future business prospects. In some cases, you can identify these forward-looking statements by the use of words such as “outlook,” “guidance,” “believes,” “expects,” “potential,” “continues,” “may,” “will,” “should,” “could,” “seeks,” “predicts,” “intends,” “plans,” “estimates,” “anticipates” or the negative version of these words or other comparable words. Such forward-looking statements are subject to various risks and uncertainties including, among other things: our ability to achieve or sustain profitability in the future; our ability to demonstrate to spine surgeons the merits of our products; pricing pressures and our ability to compete effectively generally; collaboration and consolidation in hospital purchasing; in adequate coverage and reimbursement for our products from third-party payors; lack of long-term clinical data supporting the safety and efficacy of our products; dependence on a limited number of third-party suppliers; our ability to maintain and expand our network of direct sales employees, independent sales agencies and international distributors and their level of sales or distribution activity with respect to our products; proliferation of physician-owned distributorships in the industry; decline in the sale of certain key products; loss of key personnel; our ability to enhance our product offerings through research and development; our ability to manage expected growth; our ability to successfully acquire or invest in new or complementary businesses, products or technologies; our ability to educate surgeons on the safe and appropriate use of our products; costs associated with high levels of inventory; impairment of our goodwill and intangible assets; disruptions in our main facility or information technology systems; our ability to ship a sufficient number of our products to meet demand; our ability to strengthen our brand; fluctuations in insurance cost and availability; our ability to comply with extensive governmental regulation within the United States and foreign jurisdictions; our ability to maintain or obtain regulatory approvals and clearances within the United States and foreign jurisdictions; voluntary corrective actions by us or our distribution or other business partners or agency enforcement actions; recalls or serious safety issues with our products; enforcement actions by regulatory agencies for improper marketing or promotion; misuse or off-label use of our products; delays or failures in clinical trials and results of clinical trials; legal restrictions on our procurement, use, processing, manufacturing or distribution of allograft bone tissue; negative publicity concerning methods of tissue recovery and screening of donor tissue; costs and liabilities relating to environmental laws and regulations; our failure or the failure of our agents to comply with fraud and abuse laws; U.S. legislative or Food and Drug Administration regulatory reforms; adverse effects of medical device tax provisions; potential tax changes in jurisdictions in which we conduct business; our ability to generate significant sales; potential fluctuations in sales volumes and our results of operations over the course of the year; uncertainty in future capital needs and availability of capital to meet our needs; our level of indebtedness and the availability of borrowings under our credit facility; restrictive covenants and the impact of other provisions in the indenture governing our convertible senior notes and our credit facility; continuing worldwide economic instability; our ability to protect our intellectual property rights; patent litigation and product liability lawsuits; damages relating to trade secrets or non-competition or non-solicitation agreements; risks associated with operating internationally; fluctuations in foreign currency exchange rates; our ability to comply with the Foreign Corrupt Practices Act and similar laws; increased costs and additional regulations and requirements as a result of being a public company; our ability to implement and maintain effective internal control over financial reporting; our lack of current plans to pay cash dividends; our ability to take advantage of certain reduced disclosure requirements and exemptions as a result of being an emerging growth company; potential dilution by the future issuances of additional common stock in connection with our incentive plans, acquisitions or otherwise; anti-takeover provisions in our organizational documents and our ability to issue preferred stock without shareholder approval; potential limits on our ability to use our net operating loss carryforwards; and other risks and uncertainties, including those described under the section entitled “Risk Factors” in our most recent Annual Report on Form 10-K filed with the SEC, as such factors may be updated from time to time in our periodic filings with the SEC, which are accessible on the SEC’s website at www.sec.gov. Accordingly, there are or will be important factors that could cause actual outcomes or results to differ materially from those indicated in these statements. These factors should not be construed as exhaustive and should be read in conjunction with the other cautionary statements that are included in this release and our filings with the SEC.
We operate in a very competitive and challenging environment. New risks and uncertainties emerge from time to time, and it is not possible for us to predict all risks and uncertainties that could have an impact on the forward-looking statements contained in this release. We cannot assure you that the results, events and circumstances reflected in the forward-looking statements will be achieved or occur, and actual results, events or circumstances could differ materially from those described in the forward-looking statements.
The forward-looking statements made in this press release relate only to events as of the date on which the statements are made. We undertake no obligation to publicly update or review any forward-looking statement, whether as a result of new information, future developments or otherwise, except as required by law. We may not actually achieve the plans, intentions or expectations disclosed in our forward-looking statements and you should not place undue reliance on our forward-looking statements. Unless specifically stated otherwise, our forward-looking statements do not reflect the potential impact of any future acquisitions, mergers, dispositions, joint ventures, investments or other strategic transactions we may make.
Media Contact:
Zeno Group on behalf of K2M Group Holdings, Inc.
Christian Emering, 212-299-8985
Christian.Emering@ZenoGroup.com
Investor Contact:
Westwicke Partners on behalf of K2M Group Holdings, Inc.
Mike Piccinino, CFA, 443-213-0500
K2M@westwicke.com